MOLECULAR MODELLING
The subject of research is the study of physicochemical processes at scales from angstrom to nanometers, employing and assembling different methods of analysis.
QUANTUM MECHANICS
This methodology allows us to study processes at molecular level, using the density functional theory (DFT). DFT estimates the system wavefunction, as a result different physical properties of the system could be obtained. In this line, we study surface reactivity by calculating the adsorption energy and activation energy of molecules upon it, at solid/liquid and solid/gas interfaces. Also we model the surface functionalization in order to represent the physicochemistry present in materials synthetized at the laboratory or in order to propose models for unknown material structures.
We perform parallel calculations using the Quantum Espresso (www.quantum-espresso.org), installed in the institutional cluster TUPAC.
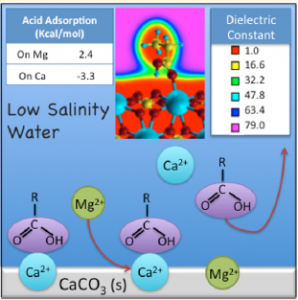 This methodology could be applied to a huge variety of scientific problems, in particular it is possible to be used to study processes related to the enhanced oil recovery. (J. Phys. Chem. C 2014, 118, 19180-19187) (J. Phys. Chem. C 2014, 118, 19180-19187).
This methodology could be applied to a huge variety of scientific problems, in particular it is possible to be used to study processes related to the enhanced oil recovery. (J. Phys. Chem. C 2014, 118, 19180-19187) (J. Phys. Chem. C 2014, 118, 19180-19187).
CLASSIC MECHANICS
This methodology allows us to study processes at the nanometer scale, it employs force field parameters to accurately represent the interaction between species in the system. These force fields could be determined by quantum mechanical calculations, resulting in an integration of the two levels of methodologies. In particular, we study processes related to fluid confined in geological pores, which is in closed relation with oil industry. This could be related to enhanced oil recovery processes as well as to shale gas/shale oil.
We employ Molecular Dynamics (MD) simulations to obtain structural and dynamical properties of the confined fluids and Gran Canonical Monte Carlo (GCMC) calculations to determine the accurate densities of the fluids confined at the oil reservoir conditions.
LAMMPS (lammps.sandia.gov) is used for MD calculations and RASPA (http://dx.doi.org/10.1080/08927022.2015.1010082) for GCMC calculations. Both installed in the institutional cluster TUPAC.
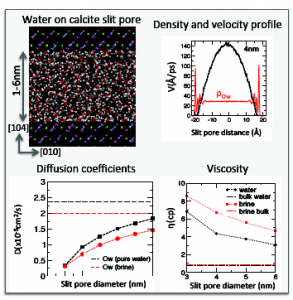 With the end of understanding the confinement effect at the carbonate reservoirs, we studied water and brine confined at calcite slit pores.
With the end of understanding the confinement effect at the carbonate reservoirs, we studied water and brine confined at calcite slit pores.
External colaborations:
Prof. Dra. Perla Balbuena (http://research.che.tamu.edu/groups/Balbuena/). Department of Chemical Engineering, Texas A&M University, College Station, TX 77843,USA
Prof. Dr. Caetano Rodrigues Miranda (http://romeo.if.usp.br/~sampasp/), Universidade de São Paulo/USP, Instituto de Física/IF, Dep. de Física dos Materiais e Mecânica/DFMT Ed. Van de Graaff – Grupo SAMPA, Rua do Matão, Travessa R, 187 – Cidade Universitária 05508-090 – São Paulo, SP - Brasil
Prof. Dr. Horacio R. Corti. Universidad de Buenos Aires, DQIAyQF-INQUIMAE, Grupo de vidrios y líquidos sobreenfriados, Pabellón 2, Ciudad Universitaria, Buenos Aires, Argetina
Prof. Dr. Damian A. Scherlis ( (http://www.inquimae.fcen.uba.ar/groups/simulation/index.html). Universidad de Buenos Aires. DQIAyQF-INQUIMAE. Laboratorio de Simulación Cuántica y Clásica en Materia Condensada a Escala Molecular. Pabellón 2, Ciudad Universitaria, Buenos Aires, Argentina